Autophagy is a series of processes in which some cytoplasmic components such as dysfunctional proteins, organelles and intracellular pathogens in the cytoplasm are transported to lysosomes for degradation and recycling of some cytoplasmic components under conditions such as nutrient deficiency, stress and pathogen infection. Autophagy not only helps to fight various diseases but also promotes the occurrence of certain diseases. When autophagy is disturbed, it will affect various human diseases including infectious diseases, autoimmune diseases, tumors, metabolic diseases, neurodegenerative diseases, cardiovascular diseases, liver diseases, lung diseases, kidney diseases, bone diseases, eye diseases and aging. More and more studies have shown that there is also a close connection between autophagy and HIV-1 infection. Autophagy can be used as an antiviral mechanism to resist invading viruses; viruses also affect autophagy during infection. Therefore, this article reviews the molecular mechanism of autophagy, the antiviral mechanism of autophagy and the impact of autophagy during HIV-1 infection, aiming to provide new ideas for achieving a possible cure for AIDS.
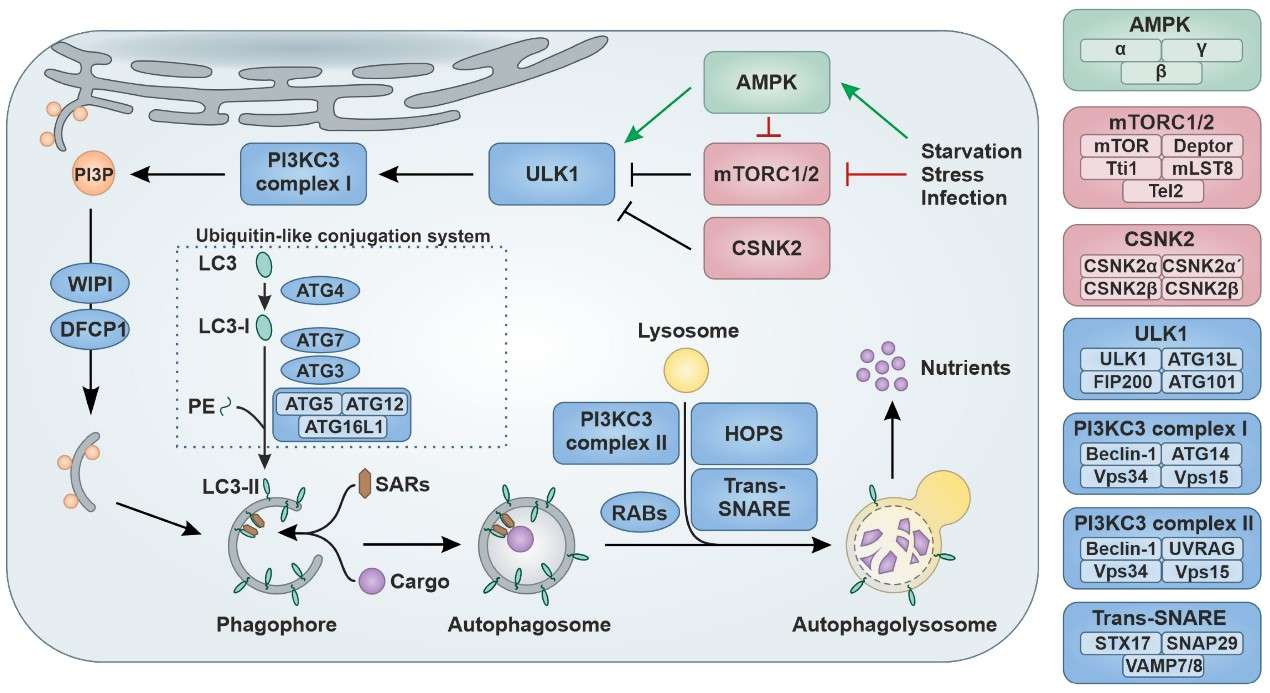 Figure 1. Schematic overview of the autophagy pathway. (Sources: Klute S, et al. 2024)
Figure 1. Schematic overview of the autophagy pathway. (Sources: Klute S, et al. 2024)
Cellular autophagy also plays an important role in the study of viral infection. Initially, after cells are infected with viruses, autophagy induces the production of interferon and triggers innate immune responses by cooperating with pattern recognition receptors (PRRs). Autophagy can also present viral antigens to T lymphocytes to initiate adaptive immune responses. Xenophagy, as selective autophagy, can specifically recognize and capture intracellular microorganisms (viruses) and pathogens in damaged intracellular vacuoles, and target them for further degradation in autophagosomes. Xenophagy depends on the recognition of invading viruses by selective autophagy receptors, which are then taken up into double-membrane autophagosomes. Finally, the ubiquitin-tagged pathogens are recognized by chelate-like receptors (SLRs) hidden in the vacuolar compartment, and the viruses are eliminated by xenophagy. In addition, during viral infection, Toll-like receptors (TLRs) induce autophagy by binding to Beclin1 through MYD88 or TRIF, which interferes with BCL-2 interaction to promote IFN production, while autophagy can negatively regulate or terminate TLR signaling by promoting the selective degradation of TRIF. Autophagy can also transmit pathogen-associated molecular patterns (PAMPs) to TLR7 or TLR8 in endosomes, promoting the production of inflammatory mediators. Viral DNA can also activate the stimulator of IFN gene protein (STING) to promote type I IFN production through the production of cGAMP synthetase and cyclic GMP-AMP (cGAMP). Removing autophagy stimulation can activate the STING pathway. Autophagy factors (such as ATG9) negatively regulate the cytoplasmic translocation of STING and inhibit the production of type I IFN mediated by TBK1. In short, after viral infection, the interaction between PRRs and autophagy positively or negatively regulates various signaling pathways in the cell, ultimately creating an optimal antiviral environment.
The early stage of HIV-1 infection includes viral adsorption and penetration and reverse transcription and integration. Among them, the viral envelope glycoprotein (encoded by the structural gene env, containing two subunits, gp120 and gp41) plays an important role in the process of viral adsorption and penetration and viral assembly and budding. When HIV-1 interacts with the main receptor CD4 and the auxiliary receptor CCR5/CXCR4 in the target cell, it mediates the entry and early infection of HIV-1. From HIV-1 infection to the development of AIDS, the core factor is the depletion of uninfected CD4 cells, and Env expressed on the surface of infected cells plays a key role in this process. Therefore, the effect of HIV-1Env on autophagy in the early stage is of great significance. Studies have shown that autophagy is inhibited in CD4 cells infected with X4 or R5 HIV-1, and autophagy is induced in monocytes/macrophages infected with X4 or R5 HIV-1. R5Env and X4Env expressed by infected cells trigger autophagy and cell death in uninfected CD4 cells but not in uninfected monocytes/macrophages, indicating that Env-triggered autophagy is cell type dependent and mediated by gp41-triggered membrane fusion, independent of co-receptors. Weaker autophagy and enhanced viral replication were observed in monocytic-differentiated macrophages (MDMs) infected with R5 HIV-1 compared with those infected with X4 HIV-1, suggesting that R5 HIV-1 is more infectious in MDMs. Recent studies have also shown that Env-mediated autophagy acts on peroxisomes to induce oxidative stress, leading to cell death in uninfected CD4 cells. Therefore, autophagy is an important antiviral process triggered by Env membrane fusion when HIV-1 infects and enters CD4 cells. Subsequently, studies have found that HIV-1Vpr also negatively regulates autophagy in the early stages of HIV-1Env infection of CD4 cells, so that autophagy is quickly controlled after HIV-1 enters the cells. In addition, HIV-1Env needs to activate ARF6-related autophagy factors to produce effective infection in CD4 cells. Since dendritic cells (DCs) are early targets of HIV-1 infection, studies have found that HIV-1Env-mediated mTOR pathway activation can inhibit autophagy in DCs, which can lead to an increase in HIV-1 in DCs and promote DC-mediated HIV-1 transfer to CD4 cells. Therefore, HIV-1 infection can activate the mTOR pathway and promote viral integration and viral replication by inhibiting autophagy.
The late stages of HIV-1 infection and replication include transcription and translation and assembly and maturation until viral release. Two regulatory genes of HIV-1 (tat transactivator and rev virion protein expression regulator) play a regulatory role in the early stages of the viral replication cycle, and four auxiliary genes (nef negative regulator, vpr viral protein r, vpu viral protein u and vif viral infection factor) play an important role in the production of the virus. Previous studies have shown the effects of viral proteins on autophagy during infection, such as transactivator (Tat) can block interferon gamma (INF-γ)-mediated autophagy in monocytes and macrophages; activation of the SRC-Akt signaling pathway inhibits autophagy in uninfected monocytes and macrophages. Negative regulator (Nef) prevents the maturation of autophagosomes in macrophages and astrocytes and hinders autophagy-mediated degradation of HIV-1; and prevents the nuclear translocation of the pro-autophagy transcription factor TFEB in macrophages. Viral infection factor (Vif) inhibits autophagy at the late stage of viral replication in CD4 cells and promotes HIV-1 replication. Viral protein r (Vpr) impedes early autophagic steps to prevent macrophage apoptosis. Viral protein u (Vpu) promotes BST2 displacement from the viral budding site through LC3-related phagocytosis. In recent years, related studies have been continuously deepened. For example, Tat also activates microglia by increasing mitochondrial damage through defective mitochondrial autophagy, while microglia exposed to Tat impair lysosomal function, leading to autophagy dysregulation and activation of NLRP3 inflammasome signaling. Tat can also induce autophagy through PELI1/K63-linked BECN1 ubiquitination, reduce ZO-1 in brain endothelial cells, and disrupt the blood-brain barrier. Tat upregulates PKM2 to activate the AKT/mTOR pathway and inhibit the AMPK pathway, thereby inhibiting autophagy and causing Tat-mediated HIV-1 transcriptional activation. Tat increases BAG3 through NF-κB signaling and induces autophagy in HIV-related neurocognitive disorder disease models. Nef can prevent the degradation of HIV-1Gag promoted by autophagy through ubiquitination through parkin/PRKN-mediated BCL2 ubiquitination and promote apoptosis. For viruses lacking Nef, continuous activation of autophagy by degradation of HIV-1Gag can effectively inhibit HIV-1 replication. ATG9A can also interact with Nef, but ATG9A promotes HIV-1 infection in a manner independent of interaction with Nef. Vpu uses the LC3C-related pathway to antagonize the antiviral ability of BST2, during which ATG5 acts as a signal bridge. In addition, under starvation conditions, HIVp17 interacts with Obg-like ATPase 1 (OLA1), destroys the OLA1-glycogen synthase kinase-3β (GSK3β) complex, leading to GSK3β hyperactivation, thereby inhibiting autophagy and ultimately enhancing the proliferation of HIV-1-infected CD4 cells.
Based on the antiviral mechanism of autophagy, the ultimate goal of the research is to explore the regulatory mechanism of autophagy in depth, develop effective therapeutic targets and effective therapeutic drugs, and transform the anti-HIV-1 effect of autophagy into clinical application. The study found that the ubiquitin-independent interaction between Tat and the p62/SQSTM1 adaptor causes Tat in CD4 cells to be degraded by selective autophagy, limiting HIV-1 infection. Methamphetamine and HIV-1Tat protein can synergistically induce microglial autophagy by activating the Nrf2/NQO1/HO-1 signaling pathway, thus providing new ideas for drug intervention in HIV-1 infected drug abuse patients. In addition, N-acetylcysteine amide can alleviate cell oxidative damage induced by HIV-1Tat and methamphetamine by correcting mTOR signaling. Nef counteracts autophagic restriction in a PRKN-dependent manner by enhancing the association between BECN1 and its inhibitor BCL2, making Nef a major autophagy antagonist in the antiviral process. TRIM5α, as a restriction factor with a combination of antiviral functions, mediates HIV-1 degradation through autophagy and proteasome-mediated mechanisms and acts as a viral sensor and effector of antiviral signaling, indicating that autophagy regulation has therapeutic potential to intervene in HIV-1 infection. PI3K/mTOR and PI3K/mTOR/BRD4 inhibitors inhibit HIV-1 replication by inducing autophagy to degrade intracellular HIV-1. Tat-BECN1, a specific autophagy activator, selectively eliminates latent HIV-1-infected memory CD4 cells in an ATG5- and ATG7-dependent manner, inhibits HIV-1 replication in human monocyte-derived macrophages (MDMs), and prevents viral infection. Trehalose can activate PIKFYVE, leading to TFEB nuclear translocation, inducing autophagy and limiting the survival of opportunistic mycobacteria during HIV-1 co-infection, which provides direction for the treatment of HIV-1 infection and its complications. Low-level ionizing radiation and latency reversal agents (LRA) can also activate latent proviruses, and then mTOR inhibitors can selectively kill HIV-1 infected cells, which also provides new ideas for combined anti-HIV-1 infection.
References
| Target | Cat. No. | Product Name | Size | Species Reactivity | Application | Detection Sample | |
| HIV | DEIA064 | Antibody to Human Immunodeficiency Virus(1+2) ELISA Kit | 96T | Human | Qualitative | serum, plasma | Inquiry |
| DEIA066 | Human Immunodeficiency Virus (1+2) Antigen and Antibody ELISA Kit | 96T | Qualitative | serum, plasma | Inquiry | ||
| DEIA2359 | Human HIV 1&2 Ag/Ab ELISA Kit | 96T | Human | Qualitative | serum, plasma | Inquiry | |
| DEIA3571 | Lentivirus Titer Kit, HIV-1 p24 ELISA | 96T | Virus | Quantitative | Tissue culture supernatants | Inquiry | |
| DEIA10155 | HIV-1 p24 ELISA Kit | 96T | Human | Quantitative | Tissue culture supernatants | Inquiry | |
| IVDEIA002 | Human anti-HIV 1+2 ELISA Kit | 96T | Qualitative | serum, plasma | Inquiry | ||
| DEIASL616 | HIV-1 gp120 Clade C ELISA Development Kit | 5 x 96T | quantitative | serum, cell culture supernatants | Inquiry | ||
| HIVEP1 | DEIA-LL152 | Human HIVEP1 ELISA Kit | 96T | Human | Quantitative | Serum, plasma, tissue homogenates and other biological fluids | Inquiry |
| HIV gp120 | ABPR-ZB198 | HIV-1 gGlycoprotein 120 Antibody Pair Set | 5 Plates, 15 Plates | HIV | sELISA | Inquiry | |
| HIV p24 | ABPR-ZB316 | HIV-1 p24/Capsid Protein p24 Antibody Pair Set | 5 Plates, 15 Plates | HIV | sELISA | Inquiry | |
| ChiVMV | DEIAPV124 | Chilli veinal mottle virus (ChiVMV) ELISA Kit | 500T/1000T/5000T | Qualitative | host plants | Inquiry |