The IPCR technique was originally introduced in 1992 by Sano et al.. The assay protocol has since been modified to improve the immobilization of the antigen and the assembly of the signal-generating immuno-complex, enabling quantitative readout and data analysis in real time.
The basic protocol of an IPCR assay includes four parts: immobilization of the antigen; assembly of the immuno-complex; signal amplification by real-time PCR; and data analysis. Immobilization of antigens can be achieved in two ways: (a) attachment of the antigen to the surface of a microplate by passive adsorption; (b) oriented immobilization of the antigen using a specific capture antibody. The immuno-complex has been assembled in four ways: (a) the biotinylated detection antibody is bridged by streptavidin to a biotinylated DNA marker; (b) the biotinylated detection antibody is tagged by an anti-biotin–DNA conjugate; (c) the detection antibody is directly conjugated with the DNA marker; (d) the detection antibody is conjugated with streptavidin, and then reacted with the biotinylated DNA marker. PCR amplification of signal has been approached in two ways: (a) the whole IPCR process is completed in the same plate. In this case, realtime PCR is carried out directly in the wells where the immunocomplex is assembled; (b) the whole IPCR is carried out in two different plates. The first plate is for assembly of the immunocomplex, and the second plate is for amplification of signal by PCR. For data analysis after the PCR, an automatic baseline correction is usually applied by the software of the instrument and the cycle thresholds (Ct) are calculated automatically. The amount of the target protein in each sample can be determined based on a standard curve plotted by the Ct values against the log concentrations of the target protein for linear correlation.
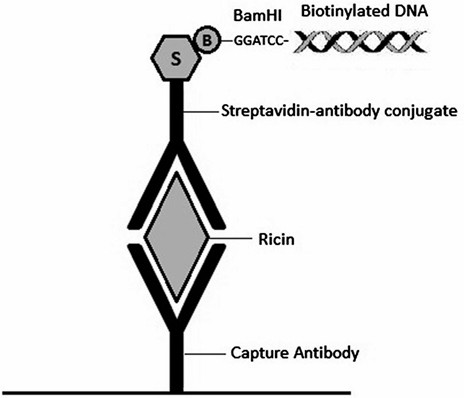 Figure 1. Schematic representation of the Sandwich IPCR method, depicting the analytical complex on the surface of an assay well.
Figure 1. Schematic representation of the Sandwich IPCR method, depicting the analytical complex on the surface of an assay well.
IPCR offers several advantages over conventional protein detection methods. First, the limit of detection is greatly improved compared to immunoassays. Second, the sample volume required is very small. The high sensitivity of IPCR enables the analysis of sample sizes of less than 1 μL. This is of particular importance for studies where only limited volumes of samples are available. Third, the assay is compatible with most complex biological matrices. Owing to the high sensitivity of IPCR, the biological sample can usually be diluted, which significantly reduces the matrix effect. Fourth, the use of real-time PCR, rather than end point PCR, improves the quantitative accuracy of the assay.
Here, we introduce a sandwich IPCR assay. This assay uses a "sandwich" of antibodies to capture and detect a protein of interest as done in a sandwich ELISA, with an additional step using a DNA marker that binds to the detection antibody through an avidin–biotin interaction allowing for signal amplification by realtime PCR.
View all ELISA Positive Control
View all ELISA Matched Antibody Pair
The method takes advantage of the high affinity bond between biotin and streptavidin to form a streptavidinated antibody-biotinylated DNA complex. Because excess proteins and DNA can interfere with PCR, the DNA is made cleavable from the antibody complex by incorporating a BamHI restriction site at the 5' end. After cleaving the DNA from the immuno-complex, an aliquot is transferred to real-time PCR tubes or 96-well plates containing a PCR master mix cocktail with primers and a probe with a fluorescent reporter. Throughout the PCR cycle, the real-time PCR cycler detects and records changes in the fluorescence signal during amplification. This amplification signal is used to calculate the cycle threshold (Ct) value. The Ct is defined as the number of cycles required for the fluorescent signal to cross the threshold (i.e., exceed background levels). Ct levels are inversely proportional to the amount of DNA in the sample such that the lower the Ct value, the greater the amount of DNA in the sample and, therefore, the greater the amount of target antigen. The DNA probe has a fluorescent reporter at one end and a quencher of fluorescence at the opposite end of the probe. The close proximity of the reporter to the quencher prevents detection of its fluorescence until the 5' to 3' exonuclease activity of the Taq polymerase breaks the reporter–quencher proximity and thus allows emission of fluorescence, which can be detected upon excitation with a laser. An increase in the product targeted by the reporter probe at each PCR cycle therefore causes a proportional increase in fluorescence due to the breakdown of the probe and release of the reporter.
As with all immunoassays, the sensitivity of IPCR largely depends on the antibodies selected to bind and detect the target antigen. The sandwich IPCR requires two antibodies, each with a strong affinity to different epitopes of the antigen. A polyclonal antibody can be used as both the capture and detection antibody but the use of a monoclonal antibody for capture tends to provide more reproducible results due to the consistent and even coating across each well. It is recommended that sandwich ELISAs be carried out to determine the best antibody pair for each particular antigen before applying the antibodies to the IPCR method. The limit of detection (LOD) for IPCR is defined as the average Ct value of the negative controls (wells containing buffer or sample matrix only without any antigen) plus three times the calculated standard deviation. In order to calculate the standard deviation and, subsequently, the LOD, it is necessary to perform the assay with triplicates for each sample.
Using the procedures described here, we demonstrate that the sandwich IPCR can detect 10 pg/mL of ricin in chicken egg and bovine milk samples and 100 pg/mL in ground beef extracts. Comparable ELISA results were in the 1–10 ng/mL range. Thus, IPCR affords sensitivity that is tenfold greater in the ground beef matrix, 100-fold greater in the milk matrix, and 1,000-fold greater in the egg matrix than the sensitivity obtained by ELISA. This IPCR is also highly compatible with complex environmental matrices. When applied to 23 environmental samples including feces, feral swine colon, soil, and water from watersheds for detecting the presence of Shiga toxin 2 produced by Shiga toxin-producing E. coli (STEC), it demonstrated a 100 % sensitivity and specificity.
Prepare all solutions using ultrapure water (prepared by purifying deionized water to attain a sensitivity of 18 MΩ cm at 25 °C) and analytical grade reagents. Prepare and store all reagents at room temperature (unless otherwise indicated). Follow all waste disposal regulations when disposing of waste materials.
Carry out all procedures at room temperature unless otherwise specified. Plate washes are carried out on an oscillating plate shaker, set to a low shaking speed (see Note 11). Wear gloves for all steps and change them frequently to avoid contamination. Keep workspace clean and free of DNA, antigens, and other sources of contamination by wiping down regularly.
Reference