
Mitochondria are a subcellular structure prevalent in eukaryotic cells and the most important source of energy in cells. Most tissue cells in the human body rely on oxidative phosphorylation of mitochondria to obtain the energy needed to maintain their metabolism. Mitochondria not only play an important role in cell signal transduction and apoptosis, but also play a decisive role in multiple metabolic pathways including the tricarboxylic acid cycle, beta oxidation, fat and cholesterol metabolism. Therefore, once mitochondrial DNA and related nuclear DNA involved in mitochondrial DNA expression are abnormal, mitochondrial diseases may be caused.
Mitochondrial respiratory chain disease
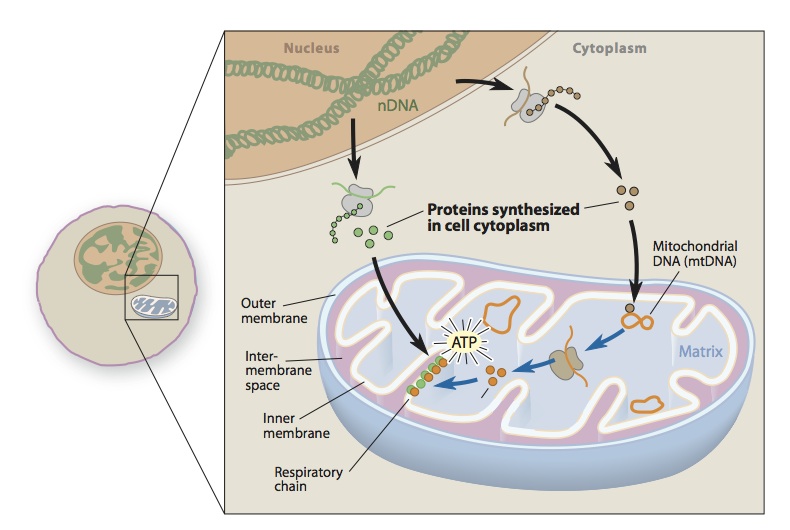
mtDNA loses more than 99% of its original genes and most autonomy in its symbiosis with nDNA. Its function depends on the basic functions of nuclear genes, including replication, translation, synthesis of most respiratory chain subunits, and assembly of respiratory chain complexes. And the synthesis of phospholipids that constitute the inner mitochondrial membrane (IMM). Mutations of respiratory chain complex subunit are induced by mitochondrial self DNA mutations and mutations in related nuclear genes. These mutations are mainly in complexes I and II, it indicates genetic mutations that are deleterious in complexes III, IV and V are rare or fatal. Most mutations in the nuclear gene of complex I or complex II subunit will cause LS, a hallmark neuropathic lesion in infancy or early childhood neurodegenerative disease. Studies have found that mtDNA mutations can also cause LS, a large number of mtDNA mutations will damage oxidative phosphorylation in the early life.
Disease caused by mitochondrial protein transport
Mitochondrial transport is a complex process involving different pathways for protein localization and each of the four gaps of mitochondria (OMM, IMM, IMS, and mitochondrial matrices). More than 1,300 protein molecules in mammalian mitochondria, only 13 are encoded by mtDNA, and the rest are encoded by nDNA, which are first synthesized in the cytoplasm and then transported to specific parts of the mitochondria. Therefore, these mitochondria proteins require a specific protein transport machine is used to complete the process. The transport machine is designed for mitochondrial input and assembly pathways of IMS proteins. It is consist with the outer membrane translocation enzyme (TOM) and endomembrane translocase (TIM), sorting and assembly machinery (SAM), and the leader sequence shift enzyme-related motor (PAM). Among them, TOM and TIM run new peptides into the corresponding gaps. Therefore, some mutations in the peptides are associated with specific enzyme defects, such as methylmalonate and pyruvate dehydrogenase complex (PDHC) deficiency. Clinical studies find some human diseases are caused by the total transport mechanism. For example, one is deafness/dystonia peptide 1 (DDP1) mutation leading to an X-linked recessive hereditary deafness, dystonia syndrome, and the other is a chaperone HSP60 mutation leading to an autosomal dominant hereditary hernia sexual paraplegia (HSP).
Disease caused by abnormal mitochondrial dynamics
This is a relatively new area of interest to clinical neurologists. Mitochondrial movement, fusion, intracellular division, and finally a tubular network may be beneficial for transporting mitochondrial to areas of high energy demand. Motor neurons with no anterior horn cells anywhere need mitochondrial movement from the cell body to the neuromuscular junction. In the process, mitochondrial dynamic processes are regulated by GTPase upstream of microtubules, kinesins, and downstream dynein. The first mitochondrial motor defect was found in a family of autosomal dominant SPG10 and kinesin-encoded gene mutations (KIF5A). Gene mutations affect regions involved in microtubule-binding proteins.
At present, there are no satisfactory treatments for diseases caused by mitochondrial DNA mutations, and most of them are limited to support symptomatic treatment, such as, anticonvulsant drugs, control of endocrine dysfunction and surgical treatment. In order to solve the problem of a large number of free radicals in patients with mitochondrial respiratory chain dysfunction, the primary treatment is to use some antioxidant drugs, such as coenzyme Q10 and N acetylcysteine. Although studies have shown that coenzyme Q10 can be effective in Parkinson’s disease and Friedreich ataxia, it lacks long-term large-scale clinical evidence. Due to the polymorphism and heterogeneity of the mitochondrial genome, gene therapy for mitochondrial diseases has emerged, such as the conversion of mutated mtDNA to normal nuclear DNA by misalignment or the correction of mitochondrial DNA mutations by introduction of a special endonuclease, etc. In addition, mitochondrial disease can be reduced through genetic counseling and prenatal testing.