
Existing research results indicate that the body’s metabolic homeostasis is closely related to body aging. The process of cellular energy production affected by metabolic stress will in turn affect the bioenergy state of the cell and the adaptability of the entire organism. In addition, studies have also found that metabolic stress in early life seems to be involved in chromatin reorganization, epigenetic changes that have a lasting impact on subsequent, and may even affect the aging process.
Studies have found that the regulatory center of these epigenetic changes is located in the mitochondria. Mitochondria not only produce a large amount of 5′-adenosine triphosphate (ATP) through tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OXPHOS) to maintain cell homeostasis, they also participate in the biosynthesis of some biological molecules, such as lipids and heme , Iron-sulfur clusters and intermediate metabolites, these intermediate metabolites can be used as signals to participate in the regulation of the rest of the cell, so that mitochondria become the center of a variety of biological processes. Through the continuous signal communication between the energy control center mitochondria and the nucleus, cells and organisms can monitor and integrate the body’s nutrient utilization and energy demand information, thereby ensuring the body’s metabolic stability.
Although the complete loss or nearly complete loss of mitochondrial function is unfavorable for cells, studies have found that partial inhibition of mitochondrial activity can promote the longevity of worms, flies, and mice. Studies on Caenorhabditis elegans indicate that in the early stages of life, the reduction of electron transport chain (ETC) activity in the mitochondria leads to extensive chromatin reorganization, which is essential for activating the mitochondrial unfolded protein response (UPRmt). This pathway can promote the restoration of mitochondrial protein homeostasis and stress. Specifically, the overall chromatin compaction induced by mitochondrial stress is manifested by the use of histone H3K9 methyltransferase SETDB1/MET-2 and its cofactors ATF7IP/LIN-65 to methylate non-essential genes to avoid the transcription of non-essential genes under stress conditions. The activation of stress-related UPRmt genes is demethylated and activated by histone H3K27 demethylases JMJD-1.2 and JMJD-3.1, thereby causing a sustained response and prolonging lifespan.
UPR mt is a transcriptional response that can induce the expression of mitochondrial chaperones and proteases, and then repair or degrade the misfolded proteins in mitochondria, and ultimately restore protein homeostasis in mitochondria. In addition, studies also found that UPR mt can also promote the reconnection of cell metabolism to reduce mitochondrial stress and promote cell survival. In Caenorhabditis elegans, when mitochondrial protein homeostasis is disrupted, UPR mt is induced, resulting in a decrease in the efficiency of mitochondrial entry, so that the transcription factor (TF) ATFS-1 that would otherwise enter the mitochondria for degradation enters the nucleus. Another TF and chromatin remodeling factor DVE-1 is also involved in UPR mt signal transduction, but unlike ATFS-1, its nuclear translocation does not depend on the efficiency of mitochondrial entry.
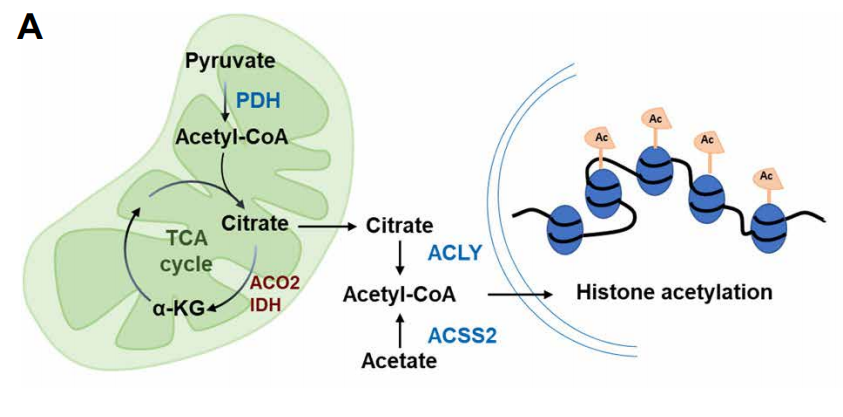
With the deepening of research, people have increasingly realized that many metabolic intermediates such as folic acid, acetyl-CoA (acetyl-CoA) and α-ketoglutarate (α-KG) not only affect the energy and material metabolism of cells , It also has a profound and dynamic impact on the overall chromatin modification. These intermediate metabolites can serve as substrates for enzymes that modify signal proteins, metabolic enzymes, and chromatin proteins. As the main source of epigenetic regulation of metabolites, such as, acetyl-CoA which is the source of histone and protein acetylation, and s-adenosylmethionine is the epigenetic methyl donor. In addition, α-KG produced in the TCA cycle of mitochondria is an essential cofactor for histone demethylase and DNA methylase. In contrast, succinic acid and fumaric acid inhibit these α-KG-dependent enzymes. In short, mitochondrial disturbances can change the nuclear epigenome by affecting various mitochondrial-derived metabolites, and ultimately affect cell proliferation and aging.
Recently, scientists have studied TF DVE-1, which is essential for UPR mt and contains homology domains, and identified nucleosome remodeling and histone deacetylases ( NuRD) complex that mediate the subcellular localization of DVE-1 in response to mitochondrial stress. The results show that after the mitochondrial function is disturbed, the level of mitochondrial-derived acetyl-CoA decreases, and the level of histone acetylation decreases, so that the NuRD complex can achieve overall chromatin reorganization. Restoring acetyl-CoA levels by providing the substrates and nutrients needed to produce acetyl-CoA can prevent the reduction of histone acetylation and inhibit chromatin reorganization under mitochondrial stress. In general, acetyl-CoA is a signal of mitochondrial dysfunction, which regulates body aging through NuRD-mediated chromatin remodeling.