
Throughout history, humans have been looking for ways to prevent aging and prolong life. With the continuous development of modern science, the process and mechanism of aging are being explained more and more. Because mitochondria endogenously generate reactive oxygen species and preferentially accumulate exogenous chemical carcinogens, mitochondrial DNA (mtDNA) is prone to mutations, leading to the accumulation of age-related somatic mutations. Clinical studies have found that some somatic mitochondrial DNA mutations in humans are pathogenic. Therefore, the theory of aging mitochondria suggests that accumulation of somatic mitochondrial DNA mutations is responsible for human aging and age-related mitochondrial respiratory defects. However, since both nuclear DNA and mitochondrial DNA are involved in encoding proteins required for mitochondrial respiratory function, there is also the possibility of age-related mitochondrial respiratory defects caused by abnormalities of nuclear DNA instead of mitochondrial DNA. Recently, Japanese researchers have discovered new information about the genetic processes that may trigger age-related diseases.
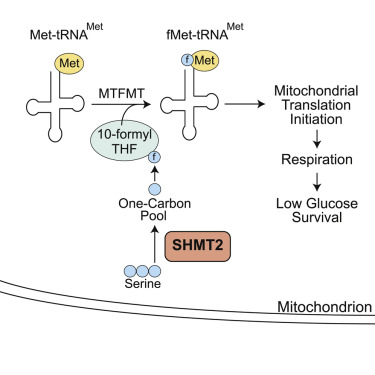
Previous research has suggested that the accumulation of somatic mutations in mitochondrial DNA (mtDNA) is the main cause of human aging and age-related mitochondrial respiratory defects. However, recent research has found another hypothesis about the mechanisms of human aging: That is, the causes of human aging and age-related respiratory deficiency diseases are due to epigenetic changes and epigenetic down-regulation of nuclear coding genes responsible for mitochondrial translation (for example, glycine C-acetyltransferase (GCAT), Serine hydroxymethyltransferase 2 (SHMT2)). To test this hypothesis, the researchers conducted studies using mouse models lacking GCAT or SHMT2 to determine whether they have a respiratory deficiency and premature aging phenotype. Experiments have shown that mice lacking Gcat do not show any macroscopic abnormalities, including premature aging phenotypes, for up to 9 months after birth. In contrast, mice lacking Shmt2 impaired mitochondrial respiration and the growth and development of liver cells. Researchers have further investigated the underlying mechanisms by which serine hydroxymethyltransferase 2 (SHMT2) gene disruption affects cellular processes such as mitochondrial respiration. They found that impaired SHMT2 expression leads to a down-regulation of the taurine-producing metabolic pathway in the liver, which in turn leads to a decrease in the concentration of taurine in liver cells. Because taurine is necessary for mitochondrial respiration, its down-regulation results in reduced cellular metabolism. This is directly related to the impairment of mitochondrial respiration and cell growth. These factors also contributed to anemia in Shmt2 knockout E13.5 embryonic mice. Since Shmt2 essentially controls the production of n-methionine-trna (fMet-tRNA) in mitochondria, its inhibition reduces mitochondrial translation, resulting in respiratory defects in embryonic fibroblasts lacking Shmt2. Therefore, age-related respiratory defects in elderly fibroblasts are not caused by mtDNA mutations, but by epigenetic regulation of nuclear genes including SHMT2. The confirmation of this hypothesis is helpful to provide a new entry point for the processes related to aging and aging diseases. It makes clinical intervention and treatment of aging and age-related diseases possible through the promotion or inhibition of genes encoding mitochondrial protein nuclear genes. This could open up new ideas for treating diseases caused by epigenetic abnormalities, and even extend lifespan.