It has long been recognized that double-stranded RNA (dsRNA) is of exogenous origin and is associated with most viral infections - it either constitutes the viral genome or is produced in the host cell during viral replication. One of the most conserved mechanisms by which cells sense viral infections is the detection of dsRNAs through a set of receptors in the innate immune system that trigger antiviral and inflammatory immune responses. However, there is growing evidence that dsRNAs are not limited to virus-infected cells, but can also be produced in different pathophysiological states by endogenous sources, such as retrotransposons and mitochondrial DNA. These endogenous nucleic acids likewise stimulate the immune system, often acting as a 'danger' signal to alert dysregulated cellular processes.
The cellular sources of dsRNAs are diverse and occur frequently in a variety of physiopathologic processes. Epigenetic derepression of transposable elements (TEs) such as endogenous retroviruses (ERVs), long interspersed nuclear elements (LINEs), and short interspersed nuclear elements (SINEs) can lead to dsRNA production. RNA post-transcriptional modifications can also regulate RNA secondary structure, and defects in mounting or recognition brought about by modifications can make themselves look like exogenous dsRNAs thereby inducing immune signals. RNA degradation mechanisms, such as those involving Dicer, the RNA ectodomain complex, and the lysosomal RNA transporter protein SIDT2, may prevent excessive accumulation of dsRNA.
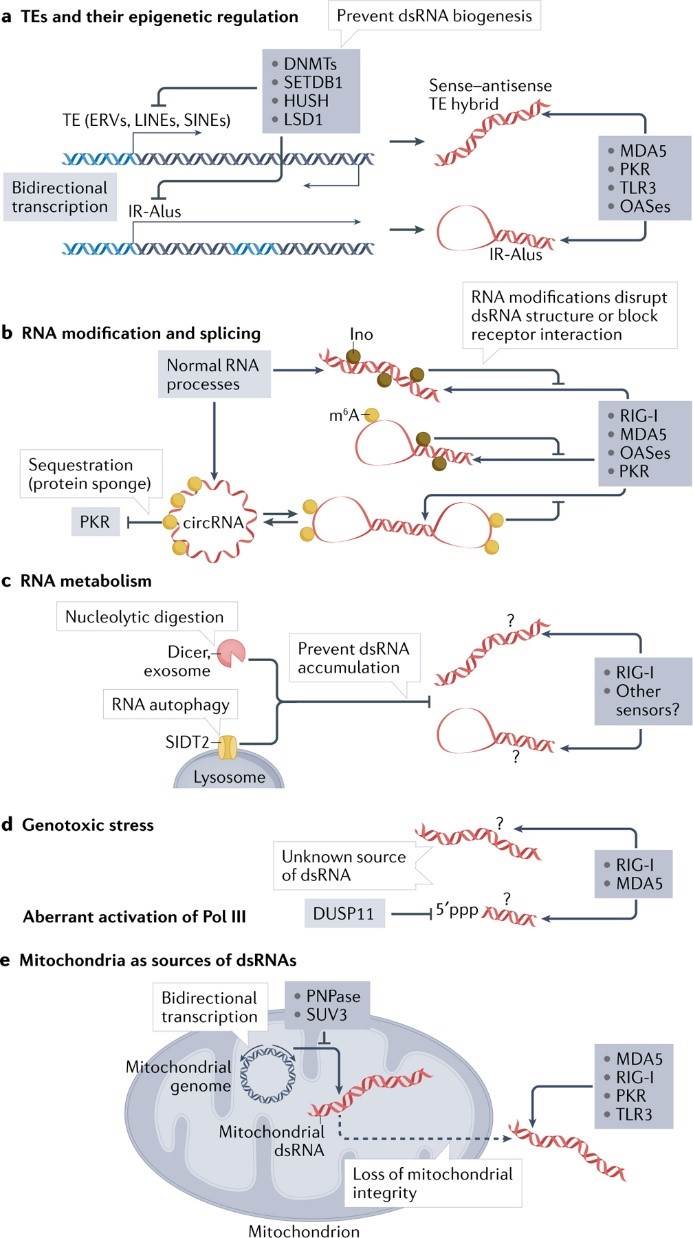 Figure 1. Endogenous sources of dsRNA and cellular regulatory processes
Figure 1. Endogenous sources of dsRNA and cellular regulatory processes
(Source: Chen YG, et al. 2022)
The structure of dsRNA is distinctly different from that of dsDNA. dsRNA displays an A-double-stranded strand with a deep and narrow major groove and a shallow and wide minor groove, while dsDNA forms a B-double-stranded strand with a wide major groove and a narrow minor groove. Thus, the primary groove of dsDNA can be used for the secondary structure of proteins, whereas the primary groove of dsRNA usually cannot. dsRNA-binding proteins interact primarily with the RNA backbone and minor grooves, indirectly affecting protein binding to some extent, but the sequence dependence of dsRNA-binding proteins is generally limited.
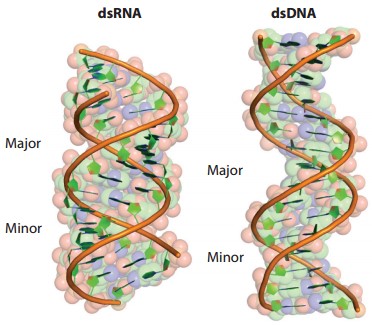 Figure 2. Structures of dsRNA and dsDNA
Figure 2. Structures of dsRNA and dsDNA
(Source: Hur S. 2019)
The immune response to viral infection often begins when viral dsRNA is detected by dsRNA-binding proteins in the host. These sensors include RIG-I-like receptors (RLRs), protein kinase R (PKR), oligoadenylate synthases (OASes), Toll-like receptors (TLRs) and NOD-, LRR- and pyrin domain-containing 1 (NLRP1).
Table 1. dsRNA-binding proteins
| dsRNA-binding proteins | dsRNA-binding domains | Best-known functions | Effect on other dsRNA-binding proteins |
| RLRs | Helicase domain, C-terminal domain | Antiviral signal activation | Induce PKR, OASes, and ADARs through interferon signaling |
| PKR | dsRBDs | Translational suppression | Amplifes RLR signaling |
| OASes, RNase L | NTase domains | Translational suppression | Amplify RLR signaling |
| ADARs | dsRBDs | RNA editing and resolving dsRNA structure | Suppress RLRs, PKR, OASes, assist Dicer |
| Dicer, Drosha | PAZ, dsRBDs, RNase Ⅲ, helicase domain | Processing dsRNAs | Inhibit RLRs |
| PACT, TRBP | dsRBDs | Regulate PKR, Dicer and RLRs | Inhibit PKR, promote Dicer and RLRs |
(Source: Hur S. 2019)
These sensors are broadly expressed in a wide array of tissues, including airway, gut and reproductive tract epithelial cells, which are primary entry sites for many viruses. Upon binding to dsRNA, these sensors activate a multitude of immune responses, including activation of antiviral and inflammatory signalling pathways, cell growth inhibition and in some cases cell death, which altogether inhibit viral replication.
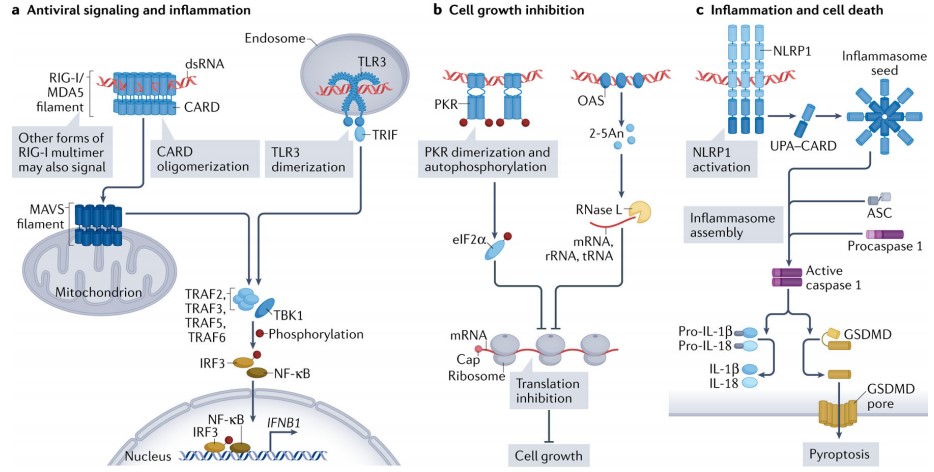 Figure 3. dsRNA binding receptor and their signalling
Figure 3. dsRNA binding receptor and their signalling
(Source: Chen YG, et al. 2022)
The RLR consists of three proteins, RIG-I, MDA5, and LGP2, all of which share similar RNA-binding structural domains, including the conserved DExD/H helicase domain. Upon recognition of viral dsRNA, RIG-I and MDA5 activate their shared downstream adaptor molecule, mitochondrial antiviral signaling protein (MAVS). Activated MAVS then recruits a variety of signaling molecules, including TNF receptor-associated factors (TRAFs), TANK-binding kinase 1 (TBK1), and interferon regulatory factor 3/7 (IRF3/7), which induces the transcription of type I interferons and other pro-inflammatory cytokines. Unlike RIG-I and MDA5, LGP2 does not directly activate MAVS but is thought to positively and negatively regulate MDA5 and RIG-I.
RIG-I recognizes dsRNA structure in a manner dependent on the dsRNA end structure, in particular the blunt end and 5' -triphosphate group (5' ppp). 5' ppp is present in all nascent transcripts but is generally removed from cellular RNAs through normal 5' processing (e.g., endoribonuclease cleavage and 7-methyl guanosine cap addition) in the nucleus. In contrast, MDA5 recognition of dsRNAs is not related to 5' ppp or dsRNA end structure, but requires long (typically 0.5-1 kb) double-stranded somatic stems, such as replication intermediates of picornaviruses.
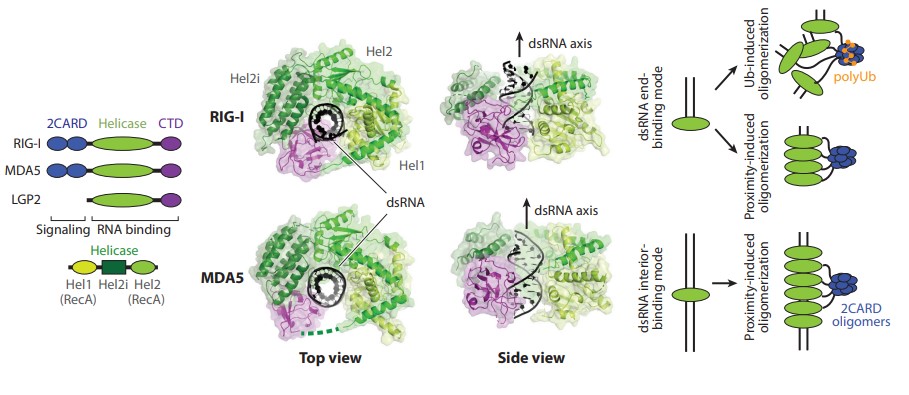 Figure 4. Domain architectures and structures of human RIG-I and MDA5 in complex with dsRNA
Figure 4. Domain architectures and structures of human RIG-I and MDA5 in complex with dsRNA
(Source: Hur S. 2019)
TLR3 is another double-stranded RNA receptor that activates antiviral signaling pathways and transcriptionally induces interferon-β and proinflammatory cytokines. Unlike cytoplasmic RLRs, TLR3 is anchored to the membrane of endosomes and detects the presence of dsRNA in the lumen of endosomes. It is generally accepted that TLR3 detects dsRNAs released from infected and dead cells by endocytosis (extracellular sensing). This is different from cytoplasmic dsRNA sensors (e.g., RLRs), which sense dsRNAs originating from within the cell (intracellular sensing). TLR3 plays a unique role in CNS host defense against HSV-1, and TLR3 deficiency results in increased susceptibility to viruses such as poliovirus and herpes simplex virus type 1 (HSV-1). The recognition of dsRNA by TLR3 also depends on the length of RNA double strands, and the minimum requirement is 40-50 bp. Nucleoside modification can inhibit TLR3 recognition, but it has also been reported that incomplete double-stranded structures in single-stranded RNA molecules can also activate TLR3, although the mechanism is not clear.
PKR is a dsRNA-dependent protein kinase that is transcriptionally upregulated by interferon, normally exists in the latent form and is activated by dsRNA binding.
One of the bestcharacterized targets of PKR is eIF2α, the α subunit of the eukaryotic translational initiation factor eIF2. Phosphorylation of eIF2α blocks eIF2 recycling and inhibits cap-dependent translation initiation. Thus, activation of PKR leads to a complete cessation of protein synthesis and inhibits viral replication, cell cycle progression, and cell growth.
PKR contains tandem repeats of two dsRNA-binding domains and a kinase structural domain.PKR activation requires stable dimerization, which can be induced by dsRNA binding and stabilized by autophosphorylation.The PKR dimeric structure has a back-to-back dimeric structure, where the two active sites are back-to-back with each other, and thus autophosphorylation may occur through inter-dimeric phosphorylation, rather than monomodal or intramolecular phosphorylation occurs. The minimum dsRNA length required for PKR activation has been reported to be ~33 bp, possibly reflecting the requirement for at least two PKR dimers to aggregate on a single RNA molecule to promote inter-dimer phosphorylation.
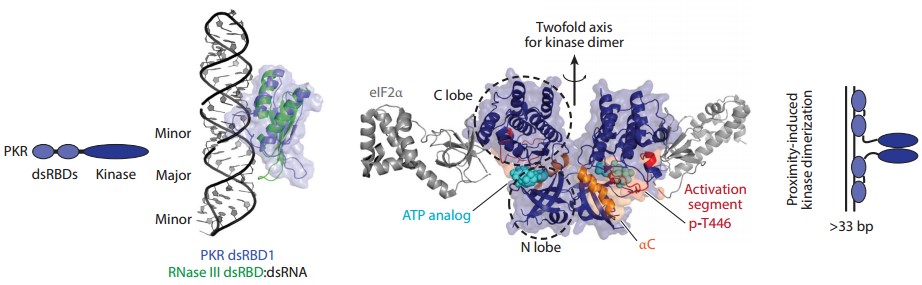 Figure 5. Domain architecture and structures of PKR
Figure 5. Domain architecture and structures of PKR
(Source: Hur S. 2019)
NLRP1 is an inflammasome-forming sensor that detects microorganisms and activates the cytokines IL-1β and IL-18 or promotes gasdermin D-mediated pore formation in the plasma membrane. Activation of NLRP1 is dependent on RNA secondary structure and RNA length, and the carboxy-terminal leucine-rich repeat (LRR) structural domain and the amino-terminal caspase 1-binding death-folding structural domain (NACHT) region on NLRP1 activate the inflammasome pathway by directly sensing dsRNAs longer than 500 bp.
The intensity of NLRP1 activation was positively correlated with dsRNA length, and it is worth noting that this activity requires a double-stranded RNA structure, and single-stranded RNAs of even longer lengths were unable to induce NLRP1 inflammasome activity. The key step in inflammasome activation is the hydrolysis of ATP by the NACHT structural domain of NLRP1 after binding dsRNA, which takes a minimum of only 15 bp dsRNA. Some of the mechanisms concerning NLPR1 are currently unknown, such as how extra length beyond 15 bp affects the inflammasome activity and antiviral response of NLRP1, the effect of base mismatches or abnormal double-stranded RNA structure on the activation of NLRP1, and whether multiple NLRP1 molecules oligomerize on a single long dsRNA.
References
| Target | Cat. No. | Product Name | Size | Species | Application | Detection Sample | |
| dsRNA | DEIA1681 | dsDNA Antibody ELISA Kit | 96T | Human | Quantitative | Human Serum or Plasma | Inquiry |
| DEIA-NS2306 | Double-stranded RNA (dsRNA) ELISA kit | 96T | N/A | Inquiry | |||
| DEIA-BZ002 | Double-stranded RNA (dsRNA, modified) ELISA kit | 96T | NA | Quantitative | Universal | Inquiry |
| Target | Cat. No. | Product Name | Host | Isotype | Application | |
| dsRNA | CABT-B212P | Mouse Anti-dsRNA Monoclonal antibody, clone sK3 | Mouse | IgG2a, κ | IP, ELISA, EM, IF, IHC | Inquiry |
| CPBT-LL016 | Mouse Anti-double-stranded RNA monoclonal antibody, clone J2 | Mouse | IgG2a, κ | Dot, ELISA, ICC, IHC, IC | Inquiry | |
| CPBT-LL017 | Mouse Anti-double-stranded RNA monoclonal antibody, clone J5 | Mouse | IgG2b | Dot, ELISA, ICC, IHC, IC | Inquiry | |
| CPBT-LL018 | Mouse Anti-double-stranded RNA monoclonal antibody, clone K1 | Mouse | IgG2a | Dot, ELISA, ICC, IHC, IC | Inquiry | |
| CABT-CS603 | Mouse Anti-dsRNA Monoclonal antibody, clone K2 | Mouse | IgM | ELISA, IHC, DB | Inquiry |