
The 2019 coronavirus disease (COVID-19) caused by the coronavirus SARS-CoV-2 is a huge challenge currently facing the global health care system. Clinical data indicate that hypoxic respiratory failure caused by acute respiratory distress syndrome (ARDS) is the leading cause of death in COVID-19 patients. Therefore, pulmonary endothelial cells (EC) have been focused on in clinical research and treatment of coronavirus diseases. The latest research shows that lung endothelial cells can affect the procoagulant state of blood by changing the integrity of the vascular barrier, and then affect the initiation and development of ARDS.
Clinical data indicate that after the initial stage of viral infection, approximately 30% of COVID-19 patients will develop severe disease with progressive lung injury. Further research found that part of the reason was caused by excessive inflammation. Through clinical studies, it was found that pulmonary complications are caused by the destruction of the vascular barrier. Damage to the vascular barrier can lead to tissue edema, endothelitis, activation of the coagulation pathway and the development of disseminated intravascular coagulation (DIC) and uncontrolled inflammatory cell infiltration.
Under normal physiological steady-state conditions, blood vessels are usually surrounded by vascular endothelial cells to maintain the integrity of blood vessels. Vascular endothelial cells control inflammation by restricting the interaction of EC immune cells and EC platelets, inhibit coagulation by expressing coagulation inhibitors and blood clot lytic enzymes, and can produce glycogenase with anticoagulant properties. In addition, recent studies have found that a subtype of pulmonary capillary ECs has a similar profile to gene expression related to antigen presentation mediated by MHC class II. Compared with ECs of other organs, the characteristics of immunomodulation of lung ECs are more obvious. These all imply that pulmonary ECs seem to be related to coagulation and excessive inflammation in ARDS.
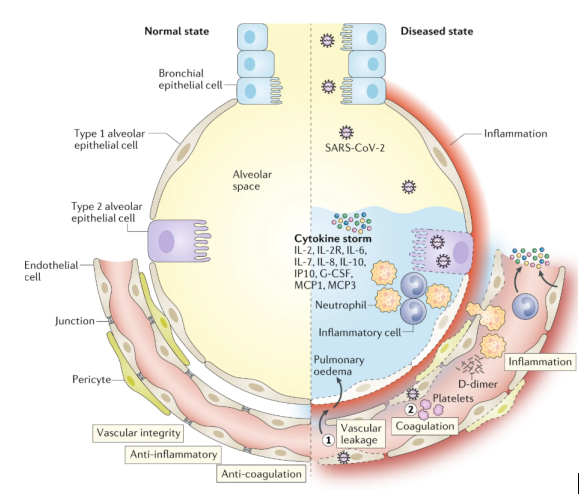
A recent clinical study on SARS-CoV-2 found that vascular leakage and pulmonary edema in severe COVID-19 patients are caused by multiple mechanisms. First, SARS-CoV-2 can directly infect the EC of multiple organs of patients. These infected ECs exhibit extensive endothelitis characterized by EC dysfunction, dissolution and death. Second, it is well known that SARS-CoV-2 binds to the ACE2 receptor, which can impair the activity of ACE2. It is worth noting that decreased ACE2 activity will indirectly activate the kallikrein-bradykinin pathway, thereby increasing vascular permeability. Third, activated neutrophils recruited to the lung EC produce tissue toxic mediators such as reactive oxygen species (ROS). Fourth, stimulation of immune cells, inflammatory cytokines and vasoactive molecules will increase EC contractility, which in turn leads to loosening of the connections between endothelium. This will cause the EC to separate, resulting in an endothelial space. Finally, the cytokines IL-1β and TNF activate glucuronidase and degrade glycocalyx, but also upregulate the expression of hyaluronan synthase, resulting in increased deposition of hyaluronic acid in the extracellular matrix and promoting Liquid retention. These mechanisms together lead to increased vascular permeability and vascular leakage.
In addition to vascular leakage, activation of the coagulation pathway is also an established feature of patients with severe COVID-19, and may further develop into disseminated intravascular coagulation. The study found that this is also related to EC activation and dysfunction. Because of the destruction of vascular integrity and the death of EC, the basement membrane of thrombosis is exposed, which in turn leads to the activation of the coagulation cascade. In addition, ECs activated by IL-1β and TNF initiate coagulation by expressing P-selectin, von Willebrand factor and fibrinogen binding to platelets. In turn, ECs release trophic cytokines, further increasing platelet production. In addition, platelets also release VEGF, which triggers ECs to up-regulate the expression of tissue factor, which is the main activating factor in the coagulation cascade. In response, the body will take appropriate measures to dissolve fibrin-rich blood clots, which explains why high levels of fibrin breakdown products can indicate a poor prognosis. Due to disseminated intravascular coagulation and blockage/congestion caused by inflammatory cells to small capillaries, and possible thrombosis in large blood vessels, lung tissue ischemic, triggering angiogenin and potential EC hyperplasia. The latter can aggravate ischemia, and angiogenesis can be used as a rescue mechanism to minimize ischemia. However, the newly formed blood vessels can also serve as channels for inflammatory cells and are attracted by activated ECs, thereby promoting inflammation.